
IBISC améliore la connaissance des ARN longs grâce à l’IA
Source(s): Genopole
Le laboratoire IBISC (Université Évry Paris-Saclay) a développé un outil d’IA permettant d’améliorer la prédiction de la structure secondaire des ARN longs et d’identifier automatiquement des sous-domaines potentiellement fonctionnels.
Le groupe de bioinformatique dirigé par Fariza Tahi au sein du laboratoire IBISC de l’Université Évry Paris-Saclay franchit une nouvelle étape dans l’analyse des ARN longs non codants, aux activités biologiques et pathologiques majeures. Les chercheurs ont développé DivideFold+. Cet outil d’apprentissage profond améliore la prédiction de la structure secondaire de ces ARN, notamment en palliant le manque de données. Il propose également une segmentation des ARN de grande taille en sous-domaines potentiellement fonctionnels, utile aux biologistes. L’outil est librement accessible à la communauté scientifique sur la plateforme logicielle génopolitaine EvryRNA.
Établir la structure pour comprendre la fonction
Les ARN jouent un rôle essentiel dans le fonctionnement des cellules. Au-delà de leur participation à la synthèse des protéines en tant qu’ARN dits « codants », de nombreux ARN non codants interviennent dans la régulation de l’expression des gènes, le développement des organismes ou encore les réponses aux variations de l’environnement. Ils sont également impliqués dans de nombreuses pathologies, notamment les cancers, les maladies auto-immunes et certaines infections virales.
Pour exercer leurs fonctions biologiques, ces molécules adoptent des structures tridimensionnelles complexes dont l’organisation repose sur une structure secondaire constituée d’appariements entre nucléotides. La détermination de cette structure est un enjeu majeur pour comprendre leur rôle biologique et identifier les régions fonctionnelles.
Cependant, si les petits ARN sont aujourd’hui relativement bien caractérisés, les ARN longs restent difficiles à étudier. Leur taille importante augmente la complexité des calculs et limite les performances des méthodes de prédiction. La présence de motifs structuraux complexes, tels que les pseudonœuds, complique encore davantage l’analyse. Les approches expérimentales sont, quant à elles, coûteuses et difficiles à mettre en œuvre à grande échelle.
Diviser pour mieux analyser les ARN longs
Pour répondre à ces défis, le jeune chercheur Loïc Omnes, sous la direction de Fariza Tahi et Eric Angel au sein de l’équipe AROB@S du laboratoire IBISC (Université Évry Paris-Saclay), a conçu DivideFold+, une version améliorée de l’outil DivideFold développé précédemment par l’équipe.
L’approche a été publiée le 18 mai 2026 dans Journal of Molecular Biology.
Le principe de DivideFold+ repose sur une approche dite « diviser pour régner » :
- les longues séquences d’ARN sont découpées en fragments plus courts et structurellement indépendants ;
- les structures secondaires de chacune sont prédites séparément avant d’être réassemblées.
Cette stratégie permet, pour les ARN de grande taille, d’améliorer à la fois la rapidité des calculs et la précision des prédictions, tout en facilitant la prise en compte de structures complexes.
Une nouvelle stratégie pour pallier le manque de données
L’une des innovations de DivideFold+ réside dans l’intégration d’une nouvelle méthode d’augmentation des données destinée à l’apprentissage profond. Les modèles d’intelligence artificielle appliqués aux ARN souffrent en effet du faible nombre de structures connues disponibles pour leur entraînement, en particulier pour les ARN longs.
Pour pallier cette limitation, les chercheurs d’IBISC ont développé une stratégie originale.
La méthode introduit des mutations, mais aussi divers événements génétiques, biologiquement possibles, comme des insertions, des délétions, des inversions, des translocations. Contrairement aux approches classiques limitées à l’augmentation du nombre de séquences, la méthode développée par l’équipe augmente aussi les données de structure secondaire en associant à chaque modification de séquence la modification de structure engendrée. Les tests réalisés montrent que cette approche améliore les performances de prédiction.
Identifier de potentiels domaines fonctionnels
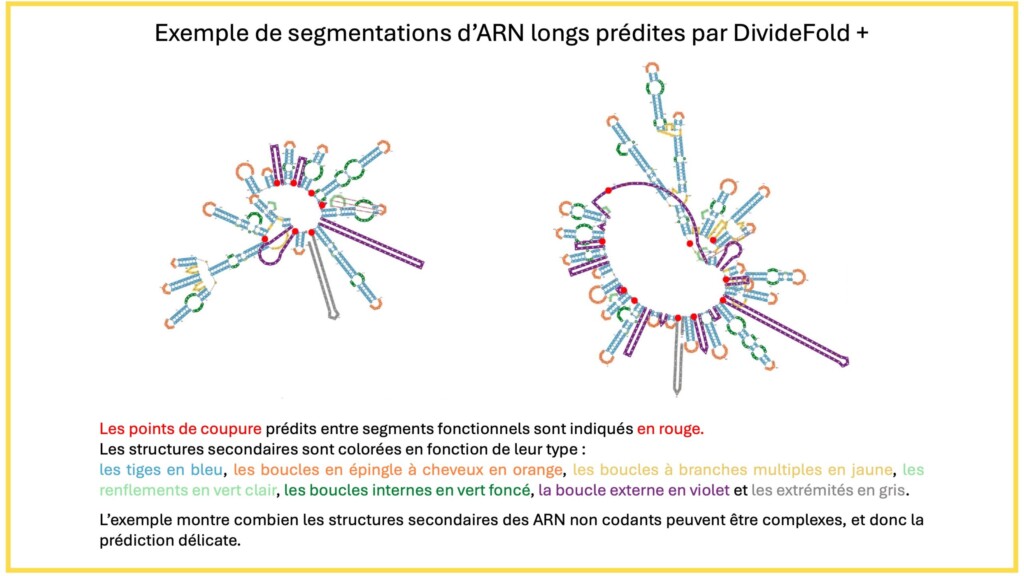
Au-delà de la prédiction structurale, DivideFold+ apporte une fonctionnalité supplémentaire : la segmentation automatique des ARN en sous-domaines (cf. figure ci-dessous). Les fragments générés lors du découpage de la séquence correspondent à des régions structurellement cohérentes qui pourraient constituer des domaines fonctionnels distincts.
DivideFold+ constitue ainsi une première étape vers l’identification automatisée de régions fonctionnelles au sein des ARN longs à partir de leur seule séquence nucléotidique. C’est une avancée particulièrement utile aux biologistes pour explorer le rôle de ces ARN dans le fonctionnement cellulaire ou le développement des maladies.
Un outil interactif accessible à tous les biologistes
À ces avancées méthodologiques s’ajoute la mise à disposition d’un serveur web interactif permettant aux biologistes de visualiser facilement la structure secondaire prédite ainsi que les différents sous-domaines identifiés. Cette interface constitue une valeur ajoutée par rapport aux outils existants, qui fournissent généralement une prédiction globale sans analyse de l’organisation interne de l’ARN.
Ainsi, avec DivideFold+, le laboratoire IBISC met à la disposition de la communauté scientifique un nouvel outil pour explorer l’architecture des ARN longs et mieux comprendre les liens entre séquence, structure et fonction. Accessible librement sur la plateforme EvryRNA, l’une des 24 plateformes mutualisées à Genopole, ce logiciel enrichit l’offre d’outils bioinformatiques développés par l’équipe pour l’étude des ARN non codants.
En conclusion
DivideFold+ est la première tentative de structuration des ARN longs en segments potentiellement fonctionnels en se basant uniquement sur leur séquence nucléotidique. Avec près de 30 outils développés, le groupe bioinformatique de l’équipe AROBAS, au sein du laboratoire IBISC, espère accélérer la connaissance dans le vaste domaine des ARN non codants, notamment sur leur implication dans les maladies, et ainsi contribuer, à terme, au développement d’approches diagnostiques et thérapeutiques.
Référence
DivideFold+: an AI-based tool for RNA secondary structure prediction with subdomains identification and visualization and data augmentation
Journal of Molecular Biology (2026)
https://doi.org/10.1016/j.jmb.2026.169865